Research Interests
Biosynthesis, regulation and structure-function of ADAMTS13 metalloprotease
Key words: Von Willebrand factor, Protease, Thrombotic Thrombocytopenic Purpura
Description of Research
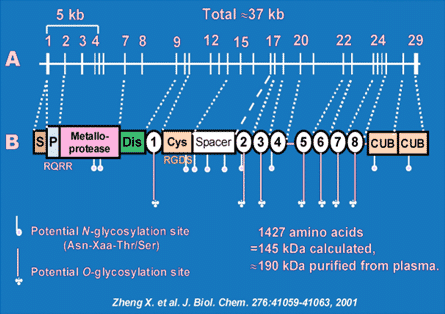
Fig. 1. Structure of ADAMTS13 gene and protein. Exons 1-29 are shown to scale above the schematic structure of the protein. Dashed lines indicate the correspondence between the boundaries of exons and the boundaries of structural domains. S, signal peptide; P, propeptide; Dis, disintegrin-like; Cys, Cys-rich. TSP1 repeats are numbered 1-8.
ADAMTS13 (A Disintegrin And Metalloprotease with ThromboSpondin type 1 repeats) is a reprolysin-like metalloprotease that limits platelet aggregation by cleavage of the Tyr1605-Met1606 bond at the central A2 subunit of von Willebrand factor (VWF). Deficiency of ADAMTS13 results in an accumulation of "unusually large" VWF multimers that are released from endothelial cells, leading to spontaneous platelet aggregation and subsequent thrombosis in small arteries, exhibiting a thrombotic thrombocytopenic purpura (TTP) syndrome. ADAMTS13 consists of a metalloprotease domain, a disintegrin domain, first thrombospondin type 1 repeat (TSP1), a Cys-rich domain and a spacer domain. The distal C-terminus contains additional 7 TSP1 repeats and 2 CUB domains (Fig. 1). Despite progresses being made over the years, the structural elements that determine substrate recongition, particularly under fluid shear stress are not well established. In addition, the regulation of ADAMTS13 function by protein- and non-protein cofactors is not known at all under physiological conditions. Therefore, we are investigating the following:
- Determine the domains of ADAMTS13 required for macromolecular substrate recognition and specificity.
- Determine the cellular origin and regulation of ADAMTS13 biosynthesis.
- Determine the cofactor-dependent regulation of ADAMTS13 protease function.
- Characterize the ADAMTS13 mutations and anti-ADAMTS13 autoantibodies in patients with acquired TTP.
- Viral vector-mediated gene therapy for phenotypic correction of hereditary Adamts13 -/- in a biological model.
Recombinant DNA, protein engineering, expression and purification, cell culture, immunofluorescent and confocal microscopy, and various other biochemical and biophysical assays will be employed in the laboratory. In addition, biological models and intravital microscopic imaging will be employed to study the anti-thrombotic effect of ADAMTS13 and variants in vivo. The advancement in this area will not only provide more insight into understanding of pathogenesis of TTP, but also offer better tools for diagnosis and therapeutic interventions for TTP syndrome and other thrombotic disorders as well.

3401 Civic Center Blvd.
Philadelphia, PA 19104
© 2024 Children's Hospital of Philadelphia. All Rights Reserved.